Lang QT-tid-syndrom
Lang QT-tid-syndrom (LQTS) er en forstyrrelse i hjertets elektriske system. Tilstanden kan arte seg som uforklarlig svimmelhet, besvimelser eller plutselig død hos ellers friske, unge personer.
Sist oppdatert:
25. apr. 2018
Innhold i artikkelen
Hva er lang QT-tid-syndrom?
Hjertet styres av elektriske signaler. Et normalt hjerteslag starter ved at et elektrisk signal dannes i det som kalles sinusknuten i høyre forkammer. Signalet brer seg fra sinusknuten til et annet knutepunkt på overgangen mellom høyre forkammer og hjertekammer (AV-knuten). Derfra brer signalet seg ut i begge hjertekamrene. På sin vei gjennom hjertet utløser det elektriske signalet sammentrekninger i hjertemuskelen. I takt med utbredelsen av det elektriske signalet starter sammentrekningene i forkamrene og avsluttes med sammentrekning av hjertekamrene. Til sammen utgjør dette ett hjerteslag.


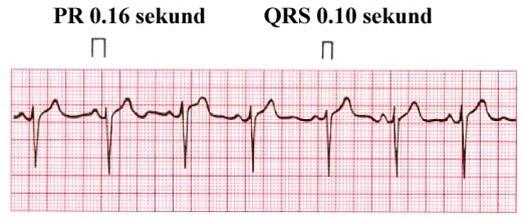
Et hjertekardiogram, EKG, viser den elektriske hjerterytmen. Hvert hjerteslag er karakterisert ved et bestemt sett av elektriske utslag på EKG'et. Det skilles mellom P-, Q-, R-, S-, T- og noen ganger U-bølger. Disse bølgene har et bestemt mønster, og de har en forholdsvis fast avstand i tid mellom hverandre.
Lang QT-tid-syndrom betyr at avstanden i tid mellom Q-bølgen og T-bølgen er for lang. Pasienter med langt QT-intervall er disponerte for rytmeforstyrrelser i hjertet. Disse rytmeforstyrrelsene kan være alvorlige, og de fører i noen tilfeller til plutselig død hos barn og yngre personer.
Tilstanden er forholdsvis sjelden, men er sannsynligvis hyppigere enn vi tidligere har trodd. Det anslås at lang QT-tid-syndrom forekommer hos 1 person per 3000-5000 individer. Tilstanden viser seg ofte med symptomer i ung barne- eller ungdomsalder.
Årsak
Lang QT-tid-syndrom er i de fleste tilfeller en arvelig tilstand, og det kan derfor være familiær opphopning av tilstanden. Den vanligste formen (Romano-Ward syndrom) arves såkalt autosomalt dominant. Det vil si at man kan få sykdom selv om kun en av foreldrene har egenskapen. En sjelden variant kalles Jervell og Lange-Nielsen syndrom. Ved dette syndromet foreligger samtidig medfødt døvhet. Totalt er det funnet 13 ulike arvelige varianter av lang QT-tid syndrom (LQT1-LQT13).
Lang QT-tid-syndrom kan også forekomme uten at det foreligger noen arvelig disposisjon (ervervet tilstand). Som regel skyldes dette bruk av legemidler som påvirker hjerterytmen. Personer som av en eller annen grunn har svært langsom puls, kan være disponerte for denne ervervede formen for lang QT-tid-syndrom.
Faren med et forlenget QT-segment er at det disponerer for anfall med en farlig hjerterytme (ventrikkeltakykardi, torsade de points). Slike anfall går i de fleste tilfellene over av seg selv, men de kan også forårsake hjertestans og død.
Symptomer
Før et hjerteanfall opptrer, er vedkommende som regel helt frisk og uten tegn til sykdom. Uten forvarsel kan et anfall inntre og arter seg vanligvis som en nesten-besvimelse, eller en besvimelse, eventuelt kun som et anfall av svimmelhet. Hjertet kan også stanse og medføre plutselig død.
Et anfall med rytmeforstyrrelse på grunn av lang QT-tid kan utløses av spenning og aktivitet, eller sterke følelsesmessige reaksjoner. Det er grunnen til at anfallene ofte melder seg under idrettsaktivitet eller trening. LQTS er en av de kjente årsakene til plutselig død hos unge idrettsutøvere.
Diagnostikk
Diagnosen stilles vanligvis etter et anfall med besvimelse, eventuelt etter vellykka gjenoppliving etter hjertestans. I noen tilfeller stilles diagnosen etter et plutselig dødsfall hos et familiemedlem, hvor utredning av andre familiemedlemmer avslører tilstanden. Hos noen stilles diagnosen etter at et tilfeldig EKG har vist QT-forlengelse.
Ved den fysiske undersøkelsen av pasienten finner legen som regel helt normale forhold. Det er EKG med måling av QT-intervallet som gir diagnosen. Diagnosen bør stilles av hjertespesialist. Vanligvis vil det samtidig bli foretatt en ultralydundersøkelse av hjertet, ekkokardiografi, for å utelukke andre sykdommer og for å lete etter eventuell underliggende hjertesykdom. For de aller fleste vil ekkokardiografien vise normale forhold.
Det anbefales å foreta en gentesting dersom man oppdager forlenget QT-tid. Utfallet av gentestingen kan ha betydning for valg av behandling, og for rådgivning i forhold til familiemedlemmer.
Behandling
Behandlingen kan deles i korttids- og langtids-strategier. Korttidsstrategien dreier seg om å få stanset et akutt anfall, det vil si å få gjenopprettet normal hjerterytme for å hindre plutselig død. Langtidsstrategien har som formål å forebygge fremtidige anfall.
Korttidsstrategien innebærer straksbehandling av ustabile rytmer (torsade de points) - som regel med hjertestarter. Det er avgjørende for utfallet av en hjertestans at de nærmeste straks setter i gang med hjerte-lungeredning inntil man får tilgang på en hjertestarter.
Ved ervervet lang QT-tid-syndrom er stans i bruken av utløsende medikament ofte det eneste som er nødvendig for å hindre nye anfall.
Alle pasienter med lang QT-tid syndrom må være nøye med å informere om tilstanden dersom det er aktuelt å bruke nye medisiner. Mange ulike medikamenter kan føre til forlenging av QT- tiden. Slike medikamenter må unngås!
Hjørnestenen i langtidsstrategien er livslang bruk av medisintypen betablokkere. Denne medisinen stabiliserer hjertet og kan forhindre anfall. I alvorlige tilfeller hvor risiko for nye episoder bedømmes å være stor, er det aktuelt å operere inn en hjertestarter og pacemaker.
Idrettsutøvere tilrås å avslutte karriæren. Siden harde fysiske anstrengelser og uttalt stress kan utløse et hjerteanfall, frarådes det å drive konkurranseidrett
Prognose
Lang QT-tid-syndrom kan føre til plutselig hjertedød hos ellers friske, unge individer. Samlet ser det ut til at ca. 6% av dem med denne tilstanden dør før de er 40 år gamle. Det er godt håp om at andelen tidlige dødsfall kan reduseres dersom tilstanden oppdages tidlig, for eksempel ved genetisk testing av familiemedlemmer til pasienter med påvist lang QT-tid.
Ekspertene antar at de fleste anfall går over av seg selv, og at bare 4-5% av anfallene fører til hjertestans og død.
Behandling med betablokker gir god prognose for de fleste. Pasienter som får operert inn en pacemaker med hjertestarter har også meget god prognose.
Dette dokumentet er basert på det profesjonelle dokumentet Lang QT-tid-syndrom . Referanselisten for dette dokumentet vises nedenfor
- Berul CI, Seslar SP, Zimetbaum PJ, Josephson ME. Acquired long QT syndrome. UpToDate, last updated May 05, 2015. UpToDate
- Chiang CE, Roden DM: The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol 2000; 36: 1-12. PubMed
- Zareba W, Moss AJ, Schwartz PJ, et al: Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med 1998; 339: 960-5. New England Journal of Medicine
- Thorsen PJ, Berg A, Hoff PI, Greve G. Risikofaktor for brå hjertedød ved lang QT-syndrom. Tidsskr Nor Lægeforen 2006; 126: 2515-9. PubMed
- Hermann Haugaa K, Berge KE, Früh A, et al. Kardiale kanalopatier - diagnostikk og behandling. Tidsskr Nor Lægeforen 2005; 125: 2778-81. PubMed
- Tranebjaerg L, Bathen J, Tyson J et al. Jervell and Lange-Nielsen syndrome: a Norwegian perspective. Am J Med Genet 1999; 89: 137-46. PubMed
- Imboden M, Swan H, Denjoy I, et al. Female predominance and transmission distortion in the long-QT syndrome. N Engl J Med 2006; 355: 2744-51. pmid:17192539 PubMed
- Moss AL. Long QT syndrome. JAMA 2003; 289: 2041-4. Journal of the American Medical Association
- Arnestad M, Crotti L, Rognum et al. Prevalence of Long-QT syndrome gene variations in sudden infant death syndrome. Circulation 2007; 115: 361-7. Circulation
- Schwartz PJ, Priori SG, Dumaine R et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med 2000; 343: 262-7. New England Journal of Medicine
- Bundgaard H, Christiansen M, Skytt Andersen P, Kjærulf Jensen H, Hastrup Svendsen J, Kjeldsen KP. Lang QT-syndrom - gener, mekanismer og risici. Ugeskr Læger 2006; 168: 2537-42. PubMed
- Berge KE, Haugaa KH, Früh A, Anfinsen OG, Gjesdal K, Siem G, Oyen N, Greve G, Carlsson A, Rognum TO, Hallerud M, Kongsgård E, Amlie JP, Leren TP. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand J Clin Lab Invest 2008; 68: 362-8. PubMed
- Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957; 54: 59-68. PubMed
- Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 2006; 113: 783-90. PubMed
- Roden DM. Acquired long QT syndromes and the risk of proarrhythmia. J Cardiovasc Electrophysiol 2000; 11: 938-40. PubMed
- Lehtonen A, Fodstad, Laitinen-Forsblom P et al. Further evidence of inherited long QT syndrome gene mutations in antiarrhythmic drug-associated torsades de pointes. Heart Rhythm 2007; 4: 603-7. PubMed
- Gussak I, Brugada P, Brugada J et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94: 99-102. PubMed
- Schimpf R, Wolpert C, Gaita F et al. Short QT syndrome. Cardiovasc Res 2005; 67: 357-66. PubMed
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20: 1391-6. PubMed
- Priori SG, Napolitano C, Giordano U et al. Brugada syndrome and sudden cardiac death in children. Lancet 2000; 355: 808-9. PubMed
- Albertella L, Crawford J, Skinner JR. Presentation and outcome of water-related events in children with long QT syndrome. Arch Dis Child 2011; 96: 704-7. PubMed
- Morita H, Wu J, Zipes DP. The QT-syndromes: long and short. Lancet 2008; 372: 750-63. PubMed
- Ali RH, Zareba W, Moss AJ, et al: Clinical and genetic variables associated with acute arousal and nonarousal-related cardiac events among subjects with long QT syndrome. Am J Cardiol 2000; 85: 457-61. PubMed
- Narayan SM. T-wave alternans and the susceptibility to ventricular arrhythmias. J Am Coll Cardiol 2006; 47: 269-81..
- Zareba W, Moss AJ, le Cessie S, et al: Risk of cardiac events in family members of patients with long QT syndrome. J Am Coll Cardiol 1995; 26: 1685-91. PubMed
- Hamang A, Solberg B, Bjorvatn C, Greve G, Øyen N. Genetic counseling in congenital long QT syndrome. Tidsskr Nor Legeforen 2009; 129: 1226-9. Tidsskrift for Den norske legeforening
- Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000; 101: 616-23. Circulation
- Bathen J, Spigset O. Lang QT-tid som bivirkning - risiko for fatale arymier Tidsskr Nor Lægeforen 2000; 120: 3432-4.
- Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med 2004; 350: 1013-22. New England Journal of Medicine
- Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high ris patients affected by the long QT syndrome. Circulation 2004; 109: 1826-33. Circulation
- Tester DJ, Ackerman MJ. Sudden infant death syndrome: how significant are the cardiac channelopathies? Cardiovasc Res 2005; 67: 388-96. PubMed