Sigdcelleanemi
Sigdcelleanemi er en medfødt sykdom som forårsaker anemi, lav blodprosent. Årsaken til sykdommen er en skade i et av genene som lager hemoglobin.
Sist oppdatert:
11. mai 2018
Hva er sigdcelleanemi?
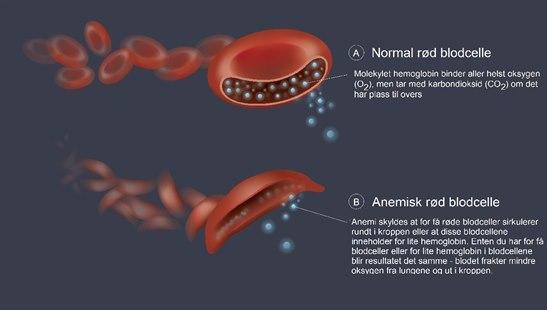
Sigdcelleanemi er en medfødt sykdom som forårsaker anemi, lav blodprosent. Årsaken til sykdommen er en skade i et av genene som styrer produksjonen av hemoglobin. Hemoglobin er det stoffet som binder oksygen til seg i de røde blodlegemene, og dermed gjør det mulig å transportere oksygen i blodet. Når hemoglobinnivået er lavt, sier vi at personen har anemi.
Sigdcelleanemi er en autosomal recessiv arvelig tilstand hvor en genfeil på kromosom nummer 6 fører til feilproduksjon av hemoglobin. Normalt hemoglobin kalles HbA. Hemoglobinet hos pasienter med sigdcelleanemi kalles HbS. HbS har en tendens til å klumpe seg inne i de røde blodcellene. På sikt kan dette føre til at blodcellene skades, og blir avlange i stedet for runde. Den avlange, litt bøyde fasongen likner på en sigd, og er årsaken til navnet sigdcelle.
For å få sykdommen sigdcelleanemi må man ha arvet ett sykdomsgen fra hver av foreldrene, slik at man har to sigdcelleanemi-gener (autosomal recessiv arvelig). Dette kalles å være homozygot. Foreldrene behøver ikke å ha vært syke - personer med kun ett gen er friske og kalles bærere av sykdomsgenet. Dette kalles å være heterozygot for denne egenskapen.
Det å værer bærer av genet er utbredt i mange land, spesielt afrikanske land og i Hellas, Italia og Midt-Østen. I USA er f.eks 8 prosent av afroamerikanere bærere av genet, men bare 1 av 400 barn av afroamerikanske foreldre har sykdommen. Man antar at årsaken til den store utbredelsen av denne genfeilen skyldes at den reduserer risiko for å bli syk eller dø av malaria.
I Norge er sigdcelleanemi sjelden, og sykdommen forekommer nesten utelukkende hos personer med afrikansk opprinnelse. Behandling er sykdommen er en oppgave for spesialister i blodsykdommer.
Animasjon om sigdcelleanemi
Symptomer
Bærere av genfeilen (heterozygote) er friske, og vil i praksis aldri få symptomer.
Pasienter som har sigdcelleanemi (homozygote), vil oppleve de første symptomene allerede i 6 måneders alder. Barna blir bleke, slappe og vokser dårlig. Alvorlige infeksjoner er vanlig. Pasienter som vokser opp, vil som regel være plaget med symptomer på anemi, f.eks. blekhet, slapphet og dårlig kondisjon. Symptomene blir mest fremtredende i forbindelse med infeksjoner og betennelser.
I ungdomsårene og voksen alder er akutte episoder på grunn av tette blodårer vanlig. Dette kan gi sterke smerter, særlig i knokler, men også i indre organer. Ofte er det nødvendig med innleggelse på sykehus under slike episoder. Et annet hyppig problem kalles akutt brystsyndrom og gir feber, hoste, tungpusthet og brystsmerter. Uten behandling kan tilstanden utvikle seg til å bli svært alvorlig.
Akutte anfall på grunn av tette blodårer kan også føre til vedvarende trange blodårer og oksygenmangel i deler av kroppen. Dette kan medføre skade av hjerne, hjerte, nyrer, milt og lever, og kan forårsake behandlingskrevende hudsår. Spesielt er risiko for hjerneslag betydelig økt blant barn med sigdcelleanemi.
Diagnostikk
For å stille diagnosen er det nødvendig å ta blodprøver. I land med høy forekomst av sykdommen er det vanlig at det gjøres screening ved fødsel. Diagnosen bekreftes i blodprøver hvor man påviser det syke hemoglobinet (HbS). Genfeilen påvises i DNA-test.
Foreldre som har stor risiko for å få barn med sigdcelleanemi, kan få utført fostervannsprøve, som med stor sikkerhet fastslår om barnet vil ha alvorlig sykdom eller ikke. Dette er særlig aktuelt for foreldre som vet at de er bærere av sykdomsgenet, dersom det er mange som har sigdcelleanemi i familien eller dersom de tidligere har født barn med sykdommen.
Behandling
Personer som er bærere av genfeilen behøver ingen behandling eller oppfølging. Det advares riktig nok mot ekstreme utmattelser i store høyder (uttørring og lite oksygen) på grunn av en teoretisk økt risiko for å få tette blodårer. Utenom dette kan bærere leve som alle andre.
Den eneste behandlingen som kan helbrede sigdcelleanemi er beinmargstransplantasjon. Dette er krevende behandling, som først og fremst er et tilbud til barn med sykdommen. Også voksne behandles i økende grad med transplantasjon dersom det finnes ressurser til det.
For de som ikke kan transplanteres består behandlingen av kontroller og oppfølging. Mange behandles med gjentatte blodoverføringer for å skifte ut mest mulig av de syke røde blodcellene. I tillegg finnes det ulike medisiner som kan lindre akutte anfall, og til en viss grad redusere antall og alvorlighet av anfallene. Mest brukt er medikamentet hydroksyurea.
Ellers er det viktig å unngå infeksjoner, uttørring, kulde og stress. Rikelig væskeinntak er viktig.
Pasienter med sigdcelleanemi følges av spesialister i blodsykdommer. Det er nødvendig med tett oppfølging og god behandling av akutte episoder.
Prognose
Bærere av ett sykdomsgen kan forvente å leve et normalt liv. Homozygot sigdcelleanemi er en alvorlig sykdom. Dersom beinmargstransplantasjon ikke er tilgjengelig eller mulig, er forventet levetid for de med alvorlig sykdom betydelig forkortet.
Dette dokumentet er basert på det profesjonelle dokumentet Sigdcelleanemi . Referanselisten for dette dokumentet vises nedenfor
- Yawn BP, John-Sowah J. Management of sickle cell disease: recommendations from the 2014 expert panel report. Am Fam Physician. 2015 Dec 15;92(12):1069-1076A.
- Stuart MJ, Nagel RI. Sickle-cell disease. Lancet 2004; 364: 1343-60. PubMed
- Mehta SR, Afenyi-Annan A, Lottenberg R. Opportunities to improve outcomes in sickle cell disease. Am Fam Physician 2006; 74: 303-10. PubMed
- Græsdal K, Gundersen K, Holm B, Waage A. Talassemi og sigdcellesykdom i Norge. Tidsskr Nor Lægeforen 2001; 121: 678-80. Tidsskrift for Den norske legeforening
- Nagel RL. Malaria and hemoglobinopathies. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, eds. Disorders of hemoglobin: genetics, pathophysiology, clinical management. Cambridge, UK: Cambridge University Press, 2001.
- Connes P, Lamarre Y, Waltz X, et al. Haemolysis and abnormal haemorheology in sickle cell anaemia. Br J Haematol. 2014;165(4):564–572.
- Gladwin MT, Rodgers GP. Pathogenesis and treatment of acute chest syndrome of sickle-cell anemia. The Lancet 2000; 355: 1476-8. The Lancet
- Roberts L, O'Driscoll S, Dick MC, et al. Stroke prevention in the young child with sickle cell anaemia. Ann Hematol 2009; 88: 943-6. PubMed
- Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148(2):94–101.
- Mantadakis E, Ewalt DH, Cavender JD, Rogers ZR, Buchanan GR. Outpatient penile aspiration and epinephrine irrigation for young patients with sickle cell anemia and prolonged priapism. Blood 2000; 95: 78-82. Blood
- Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for management of the acute painful crisis in sickle cell disease. Br J Haematol 2003; 120: 744-52. PubMed
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639-44. New England Journal of Medicine
- Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med 2000; 342: 1855-65. New England Journal of Medicine
- Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: a review. Int J Infect Dis. 2010;14(1):e2–e12.
- Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014; 312:48. doi: 10.1001/jama.2014.7192 DOI
- Adams RJ, Brambilla D; Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005; 353: 2769-78. PubMed
- DeBaun MR, Gordon M, McKinstry RC, et al. Controlled Trial of Transfusions for Silent Cerebral Infarcts in Sickle Cell Anemia. N Engl J Med 2014; 371: 699-710. doi:10.1056/NEJMoa1401731 DOI
- Hirst C, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal infection in children with sickle cell disease. Cochrane Database of Systematic Reviews 2014, Issue 11. Art. No.: CD003427. DOI: 10.1002/14651858.CD003427.pub3. DOI
- Abrams DI, Couey P, Dixit N, et al. Effect of Inhaled Cannabis for Pain in Adults With Sickle Cell Disease: A Randomized Clinical Trial. JAMA Netw Open. 2020 Jul 1;3(7):e2010874. PMID: 32678452 PubMed
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011; 377: 1663-72. PubMed
- Stettler N, McKiernan CM, Melin C, et al. Proportion of adults with sickle cell anemia and pain crises receiving hydroxyurea. JAMA 2015; 313: 1671. doi:10.1001/jama.2015.3075 DOI
- U.S Food and drug administration. FDA approves new treatment for sickle celle disease. FDA News release 2017. www.fda.gov
- Vichinsky E. New therapies in sickle cell disease. The Lancet 2002; 360: 629-31. The Lancet
- Gladwin MT, Kato GJ, Weiner D et. al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA 2011; 305: 893-902. Journal of the American Medical Association
- Sasongko TH, Nagalla S, Ballas SK. Angiotensin-converting enzyme (ACE) inhibitors for proteinuria and microalbuminuria in people with sickle cell disease. Cochrane Database Syst Rev 2015; 6: CD009191. pmid:26041152 PubMed
- Badawy SM, Payne AB. Association between clinical outcomes and metformin use in adults with sickle cell disease and diabetes mellitus. Blood Adv 2019; 3: 3297-306. pmid:31698459 PubMed
- Vichinsky E, Hoppe CC, Ataga KI, et al . A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med 2019. pmid:31199090 PubMed
- Hirst C, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal infection in children with sickle cell disease. Cochrane Database of Systematic Reviews 2012, Issue 9. Art. No.: CD003427. DOI: 10.1002/14651858.CD003427.pub2. DOI
- Ferster A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88:1960-1964. Blood
- Charache S, Terrin M, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crisis in sickle cell anemia. N Engl J Med 1995;332:1317-1322. New England Journal of Medicine
- Oniyangi O, Omari AAA. Malaria chemoprophylaxis in sickle cell disease. Cochrane Database of Systematic Reviews 2006, Issue 4. Art. No.: CD003489. DOI: 10.1002/14651858.CD003489.pub2. DOI
- Cohen AR, Norris CF, Smith-Whitley K. Transfusion therapy for sickle cell disease. In: Capon SM, Chambers LA, eds. New directions in pediatric hematology. Bethesda MD: American Association of Blood Banks, 1996:39-85.
- Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998; 91: 288-94. Blood
- Platt OS, Brambilla DJ, Rosse WF et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639-44. New England Journal of Medicine
- Overtuff GD, Powars D, Baraff LJ. Bacterial meningitis and septicemia in sickle cell disease. Am J Dis Child 1977;131:784-787. PubMed
- Meremikwu MM. Sickle cell disease. Clin Evid 2003; 10: 21-36. PubMed